研究 基礎研究
基礎研究:血液内科研究室
研究活動
近年、次世代シーケンス技術の発達により、様々な悪性腫瘍において遺伝子異常の全体像が明らかとなってきました。そのような流れの中で、我々は造血器腫瘍を中心として網羅的な遺伝子解析に取り組んでおり、世界に先駆けて、成人T細胞白血病リンパ腫などの遺伝子異常の全体像を解明し、その臨床的な意義を明らかにしてきました(K Kataoka et al., Nat Genet. 2015; K Kataoka et al., Blood. 2018)。さらに、その結果に基づいて、がん横断的な解析(全がん解析)を行い、免疫チェックポイント阻害剤の標的として注目されているPD-L1遺伝子のゲノム異常が様々な悪性腫瘍に存在し、がん免疫からの回避に関与することを明らかにしてきました(K Kataoka et al., Nature. 2016; K Kataoka et al., Leukemia. 2019)。また、さらに多数例の全がん解析を通じて、がん遺伝子における複数変異が相乗的にがん化を促進するという新たな発がん機構を解明しました(Y Saito, J Koya et al., Nature. 2020)。このように、我々は次世代シーケンスによりがんの遺伝子異常の全体像を解明することによって、創薬標的やバイオマーカーとなり得る新規のがん関連遺伝子を同定すること、および、同定された遺伝子異常に基づいて、分子生物学的手法や動物モデルなどを駆使することにより、がんの分子病態を理解することを目指しています。さらに、臨床(研究)と連携して同定された遺伝子異常の臨床的な意義の確立や、臨床シーケンスなどを含めた個別化医療への応用に取り組んでいます。
研究テーマ(1)
「次世代シーケンスによるがんゲノム解析」
造血器腫瘍を含むすべての悪性腫瘍は、ドライバーとなるがん関連遺伝子の機能を変化させる体細胞異常を獲得することにより引き起こされます。近年、我々のグループも含め世界中の主要ながん研究機関によって、大規模かつ系統的な遺伝子解析研究が行われ、幅広いがん腫において遺伝子異常の全体像が明らかにされつつあります。これらの成果は、シーケンス技術の発達、特に大量並列シーケンシング(次世代シーケンシング:next-generation sequencing: NGS)の開発により実現されたものであります。その結果、遺伝子変異(点突然変異や挿入・欠失変異)、コピー数異常(増幅および欠失)、構造異常(転座、欠失、逆位、重複)、融合遺伝子などの様々な種類の体細胞異常が明らかにされ、がん発症・進展に重要であるドライバー遺伝子の網羅的なカタログが生成されるだけでなく、腫瘍化に至る生物学的過程の解明が進みつつあります。さらに、大規模シーケンスの結果、古典的にがんの発症に重要と考えている、細胞の生存・増殖に関与するシグナル伝達経路の異常(RASやBCR-ABLなど)や、腫瘍抑制遺伝子(TP53やRB1など)に加えて、エピジェネティック制御因子(IDH1/2やDNMT3Aなど)やスプライシング因子(SF3B1やU2AF1など)、免疫回避関連遺伝子(PD-L1など)などの新たなドライバー遺伝子が見出されています。これらの中には、多数の潜在的な治療標的や、治療反応性や予後に影響を与える遺伝子異常(バイオマーカー)が含まれると考えられ、今後臨床への応用が進むと考えられています。
「成人T細胞白血病リンパ腫における網羅的遺伝子解析」
このような流れの中で、我々は世界に先駆けて、多数の成人T細胞白血病リンパ腫(adult T-cell leukemia/lymphoma: ATL)患者について、全エクソン解析・全ゲノム解析による変異および構造異常の検出、RNAシーケンスによる融合遺伝子の同定、マイクロアレイによるコピー数解析やDNAメチル化解析を含む、包括的な遺伝子解析を行ってきました。その結果、ATLにおける遺伝子異常の全体像を解明し、PLCG1やPRKCB、VAV1などの機能獲得型変異や、CTLA4-CD28融合遺伝子などの構造異常を含む、多数の新規の遺伝子異常を同定してきました(K Kataoka et al., Nat Genet. 2015; Y Kogure Y et al., Cancer Sci. 2017)。さらに、これらの遺伝子異常が臨床的因子とともに予後に影響を与えることを明らかにしてきました(K Kataoka et al., Blood. 2018)。
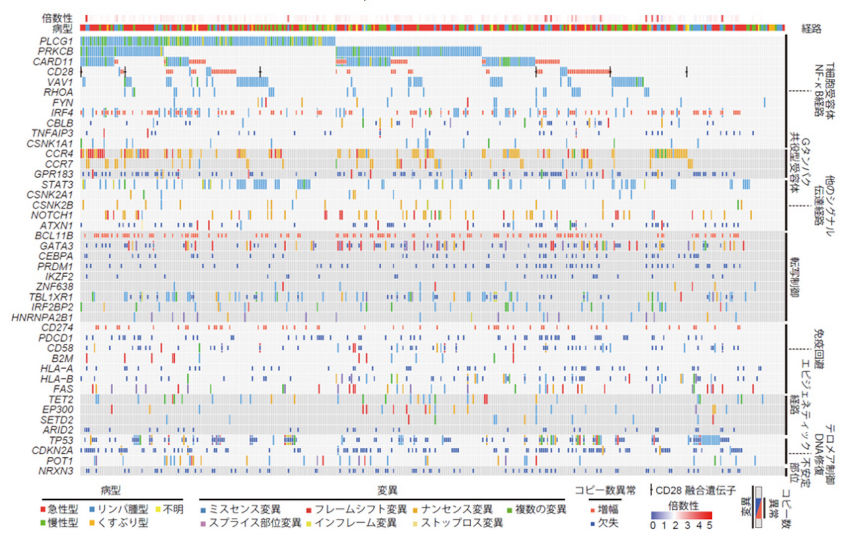
図:ATLにおける遺伝子異常の全体像(K Kataoka et al., Nat Genet. 2015)
「成人T細胞白血病リンパ腫における高深度全ゲノム解析」
これまで我々が実施してきた成人T細胞白血病リンパ腫(adult T-cell leukemia/lymphoma:
ATL)の統合解析により、多数のATLの遺伝子異常が明らかにされてきました。しかし、これまでの解析はエクソン領域を中心とした解析であり、未解明のゲノム領域が残されていました。そこで、我々は国内外の施設から150例のATLサンプルを収集し、全ゲノム解析を行いました(Kogure
Y et al., Blood. 2021.)。
タンパクコード・非コード変異、構造異常、コピー数異常のデータを統合して解析することで、56個のドライバー遺伝子を同定しました。この中には、以前は解析されていなかったエクソンに多数の異常を認めたCIC遺伝子(ATLの33%)や、構造異常によるC末端切断が高頻度に起きていたREL遺伝子(ATLの13%、GCB型のDLBCLで13%)等の11個の新規遺伝子が含まれていました。また、タンパク非コード領域ではスプライス部位の変異を繰り返し認めました。さらに、全ゲノム解析による遺伝子異常の情報を用いて、ATL患者を二群に分類でき、この分類は臨床所見や予後と関連していることを明らかにしました。このように全ゲノム解析は、様々な種類の異常を包括的に同定することができ、今後のがん研究において不可欠な技術と考えられます。また、本研究で得られた知見は難治性血液がんであるATLの新たな診断法や治療薬の開発につながる基盤となることが期待されます。
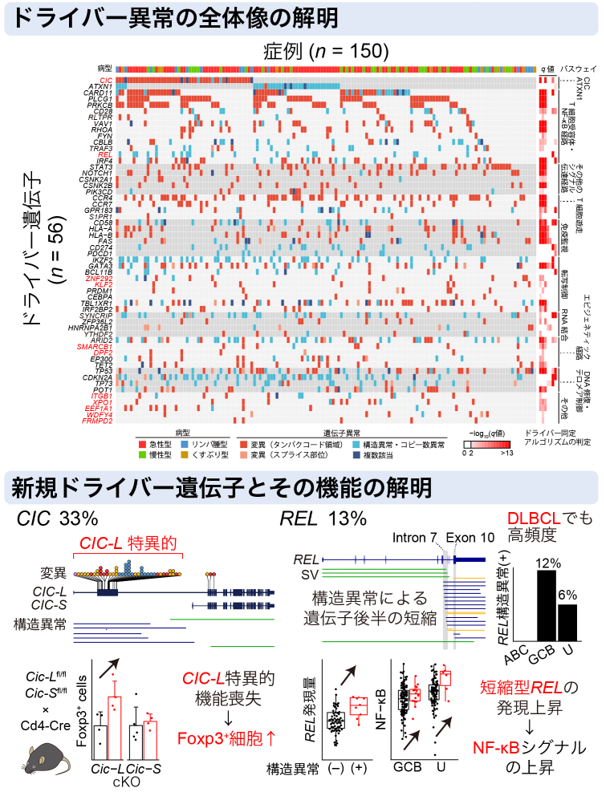
図:全ゲノム解析で解明されたATLのゲノム異常の全体像(Kogure Y et al., Blood. 2021.)
「PD-L1ゲノム異常の同定とがん免疫回避における役割の解明」
ATLにおける遺伝子解析、およびthe Cancer Genome Atlas(TCGA)で利用可能な30種類以上の悪性腫瘍における10,000例を超えるがん横断的な解析を行い、ATLと12種類の主要な悪性腫瘍(消化器がん、肺がん、頭頸部がん、B細胞性リンパ腫など)においてPD-L1 3′-非翻訳領域(untranslated region: UTR)を標的とするゲノム異常によりPD-L1の恒常的活性化が認められることを明らかにしました(K Kataoka et al., Nature. 2016)。さらに、CRISPR/Cas9システムおよびマウス腫瘍退縮モデルを用いた解析により、PD-L1 3′-UTR異常を持つ腫瘍は免疫回避による腫瘍増殖が促進されるが、その効果はPD-1/PD-L1阻害により著しく減弱されることを明らかにしました。これらの結果は、PD-L1 3′-UTR異常が免疫チェックポイント阻害剤の治療標的となり、そのバイオマーカーとして有用である可能性だけでなく、遺伝学的メカニズムが腫瘍細胞の免疫回避に重要な役割を果たすことを示しています。
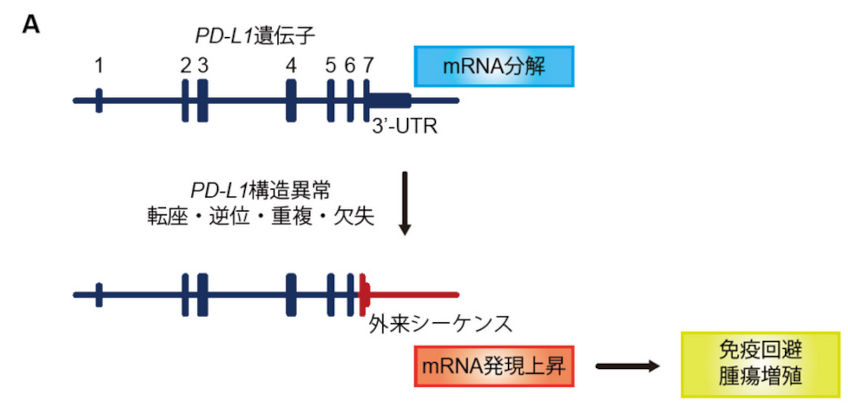
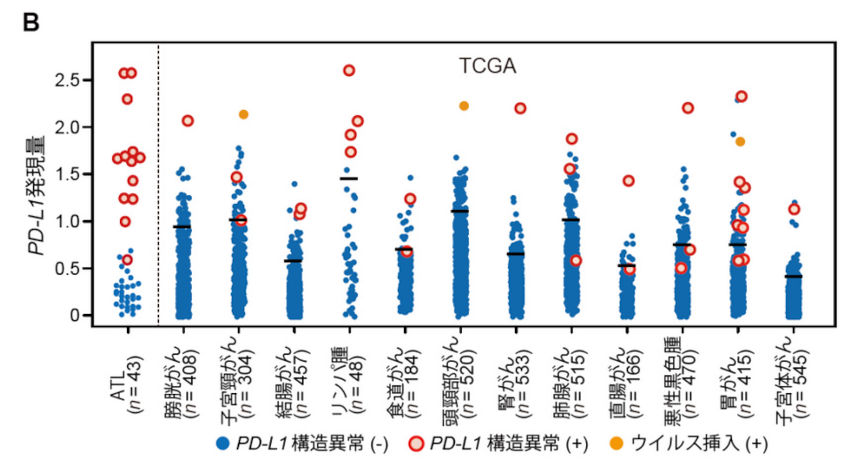
図:様々ながん腫におけるPD-L1 3′-UTR異常(K Kataoka et al., Nature. 2016)
A. 様々な種類の構造異常によりPD-L1 3′-UTR異常が起こると、mRNA分解が抑制されてPD-L1恒常的活性化が誘導され、がん細胞の免疫回避が促進される。B.
各悪性腫瘍におけるPD-L1 3′-UTR異常とPD-L1発現の関係。
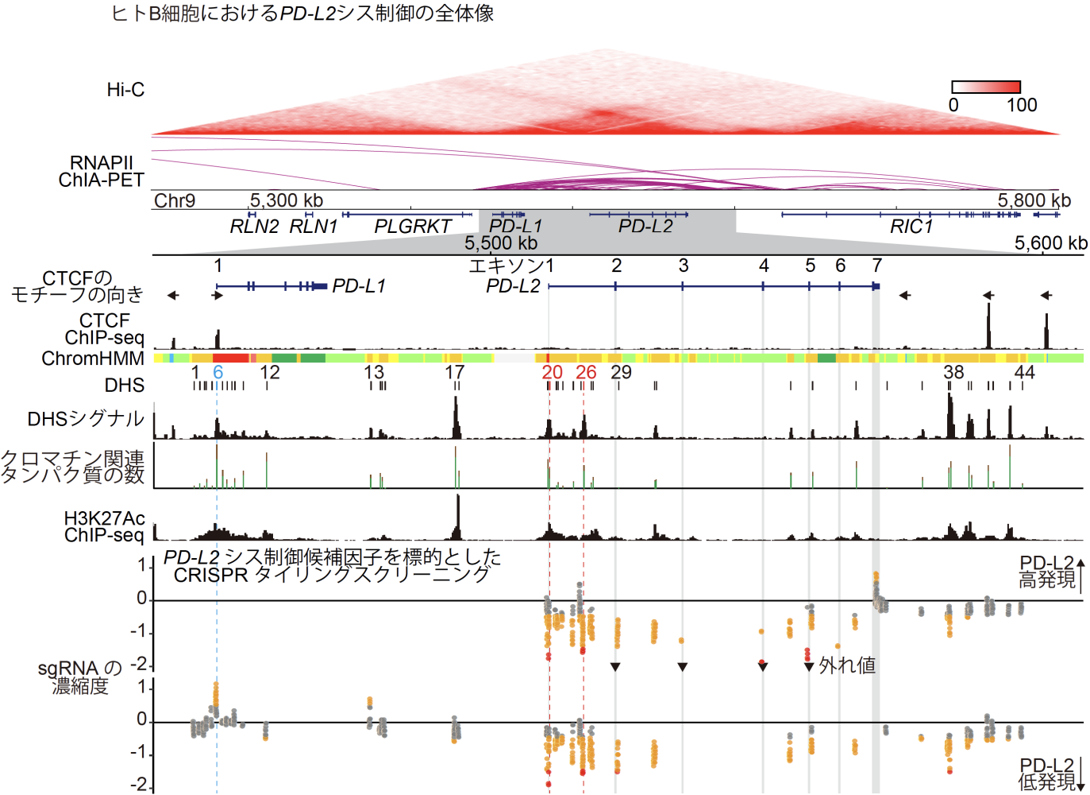
「B細胞リンパ腫におけるPD-L2の機能および発現制御機構の解明」
我々は、マウスを用いた単一細胞マルチオミクス解析と、シス制御部位およびトランス制御因子に対するCRISPRスクリーニングにより、B細胞リンパ腫におけるPD-L2の腫瘍促進機能と発現制御機構を解明しました(Shingaki S, Koya J, Yuasa M et al., Leukemia, 2022)。PD-L1とPD-L2は、免疫チェックポイント分子PD-1のリガンドです。これまで多く研究されているPD-L1とは対照的に、PD-L2のがんにおける生物学的意義と発現制御機構についてはほとんど知られていませんでした。我々は全がん解析によりPD-L2がびまん性大細胞型B細胞リンパ腫で最も高発現であることを見出し、細胞系列特異的な機能と発現制御に関する研究を実施しました。まず、B細胞リンパ腫マウスモデルを用いた単一細胞マルチオミクス解析を行い、腫瘍微小環境においてPd-l1とPd-l2の過剰発現が骨髄系細胞の増殖を促すなど、細胞動態や表現型に同等の影響を与えることを明らかにしました。次に、ヒトB細胞を用いたエピジェネティック解析とCRISPRタイリングスクリーニングの統合解析の結果、B細胞リンパ腫で特徴的に発現している新規の転写開始点を含む、複数のPD-L2シス制御部位を見出しました。さらに、機能喪失型CRISPRスクリーニングにより、IRF4とEBF1などのPD-L2トランス制御因子を網羅的に同定しました。本研究により、B細胞リンパ腫の腫瘍免疫に対する理解が進み、がん免疫療法の改善に繋がることが期待されます。当研究室は、このように大規模がんゲノムデータ解析、単一細胞マルチオミクス解析、CRISPR/Cas9システムなど、様々な研究手法を統合的に駆使することにより、がんの生物学的特性の理解に取り組んでいます。
「単一細胞マルチオミクスによるがん組織の不均一性の解明」
我々は、最新技術である単一細胞マルチオミクス解析を用いて、ヒトT細胞白血病ウイルス1型(HTLV-1)感染を原因とする成人T細胞白血病リンパ腫(ATL)の多段階発がん分子機構を解明しました。(Koya
J, Saito Y et al., Blood Cancer Discov, 2:450-467,
2021.)。単一細胞マルチオミクス解析は、同一の単一細胞から網羅的遺伝子発現のみならず100を超える細胞表面マーカー、T/B細胞受容体レパトアなどの情報を得られる最新の研究手法です。この手法を用いて、HTLV-1感染細胞を単一細胞レベルで正確に同定し、HTLV-1感染細胞のクローン拡大およびATLへの進展に伴う分子学的な細胞動態の変化を網羅的に明らかにしました。他にも、ATLクローン進展様式の多様性やHTLV-1感染やATL発症に伴う免疫微小環境動態の変化、腫瘍細胞の体細胞異常による微小環境の変容などの様々な事象が、多くの機能解析実験と組み合わせることで紐解かれました。当研究室は、この単一細胞マルチオミクス解析技術をさらに発展させ、機能解析と融合させることで、がん組織における不均一性と細胞間相互作用の理解を深化させ、新たな治療標的の探索を行います。
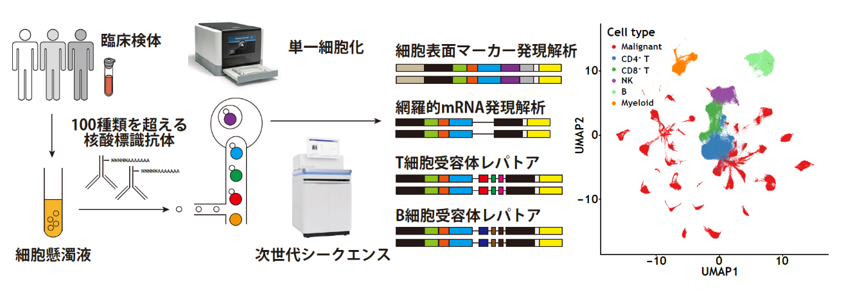
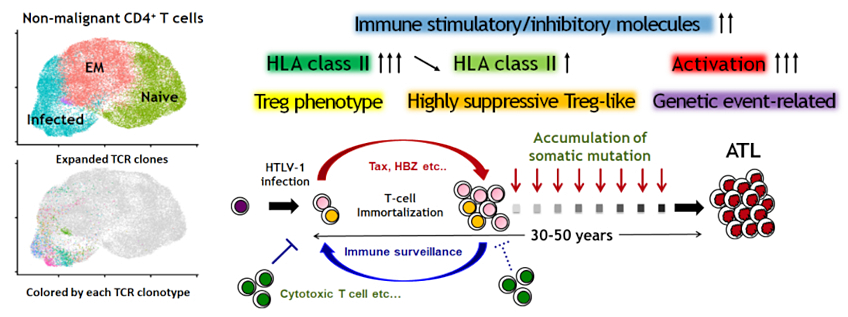
図:単一細胞マルチオミクス解析によるATLの病態解明 (Koya J, Saito Y et al., Blood Cancer Discov, 2021.)
「造血器腫瘍に対する遺伝子解析パネル検査の社会実装」
我々をはじめとして様々な研究グループから、造血器腫瘍の発症・進展に重要なドライバー遺伝子異常が報告されています。さらに、これらの遺伝子異常は、様々な造血器腫瘍において治療薬の選択だけでなく、疾患の診断や予後予測に有用であることが報告されており、遺伝子異常をまとめて検出することが患者一人一人に合わせた「個別化医療」の実現に繋がることが期待されています。しかしながら、造血器腫瘍において重要な遺伝子異常は、塩基置換、コピー数異常、構造異常、融合遺伝子と多岐に渡り、これらを1度の検査で網羅的に検査できるNGSを用いた遺伝子解析パネル検査は、日本では実装されていません。我々は日本血液学会より発表された造血器腫瘍ゲノム検査ガイドラインに基づき、造血器腫瘍において重要な遺伝子異常を網羅的に検出可能な遺伝子解析パネル検査の開発を行っています。
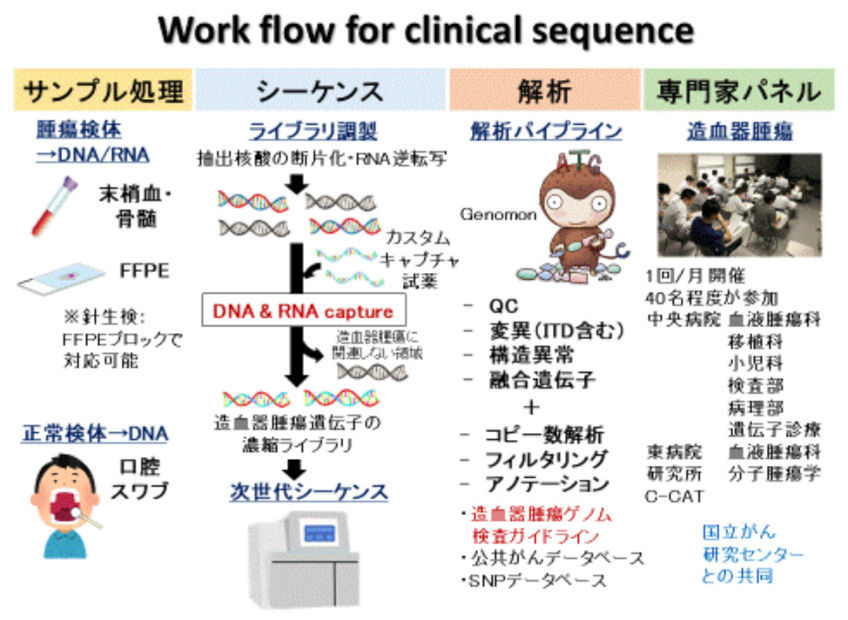
さらに、当センター中央病院・東病院、ゲノム解析実績を有する医療機関、診断薬開発企業などと共同で、この遺伝子パネル検査の性能検証および検体入手からエキスパートパネル、結果報告に至るまでの臨床シーケンスの実用性の評価を行いました。この研究には当センター中央病院・東病院で治療を受けた血液がん患者(初発および再発の成人および小児患者)が対象となり、176人の患者を解析しました。97%の患者で1個以上の遺伝子異常が、1人の患者につき中央値7個の遺伝子異常が検出されました。特にIGH転座を始めとする構造異常や、稀なものを含む様々な融合遺伝子、生殖細胞系列の異常も検出でき、本遺伝子パネル検査の高い性能が示されました。また、診断、治療法選択、および予後予測における有用性の観点から、検出された遺伝子異常を血液がんの疾患ごとに評価しました。その結果、診断、治療法選択、予後予測を行う上で臨床上有用であると考えられる遺伝子異常が、それぞれ82%、49%、58%の患者で検出され、遺伝子パネル検査は特に診断、次いで予後予測に有用であることが示されました。本研究で得られた知見は日本における血液がん遺伝子パネル検査の普及および個別化医療の基盤となることが期待されます。
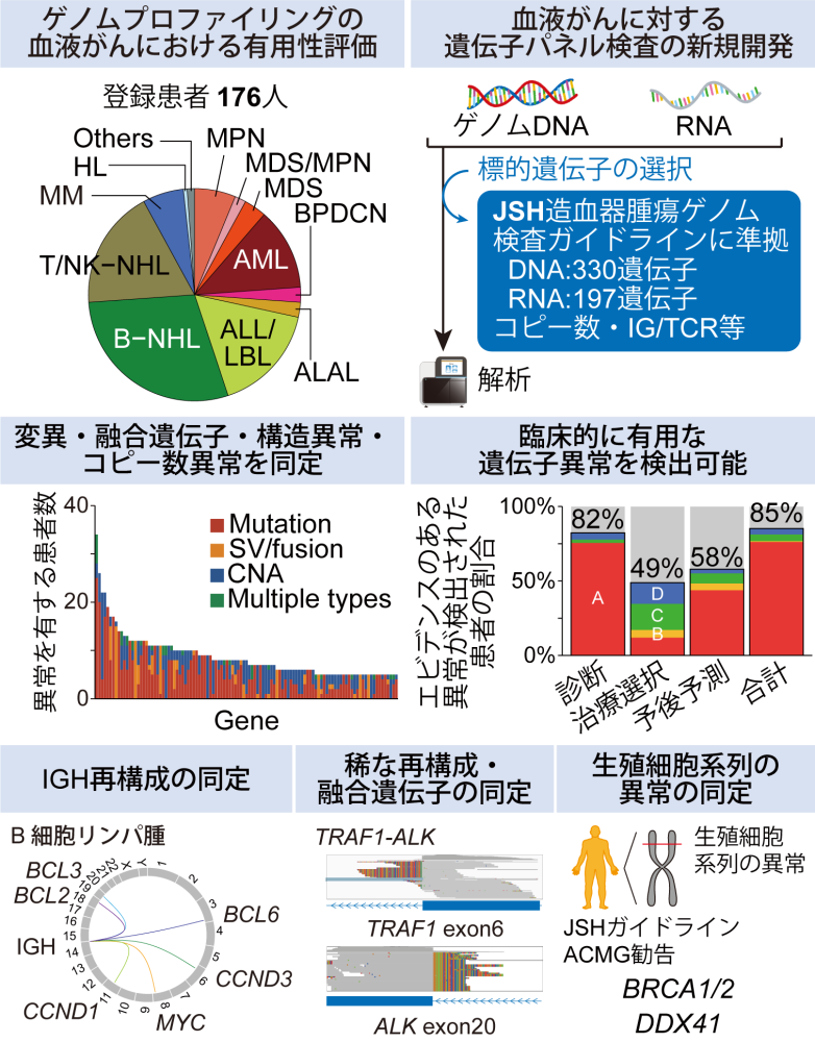
「血液がんに対する包括的ゲノムプロファイリングのための遺伝子パネル検査の有用性を検証」
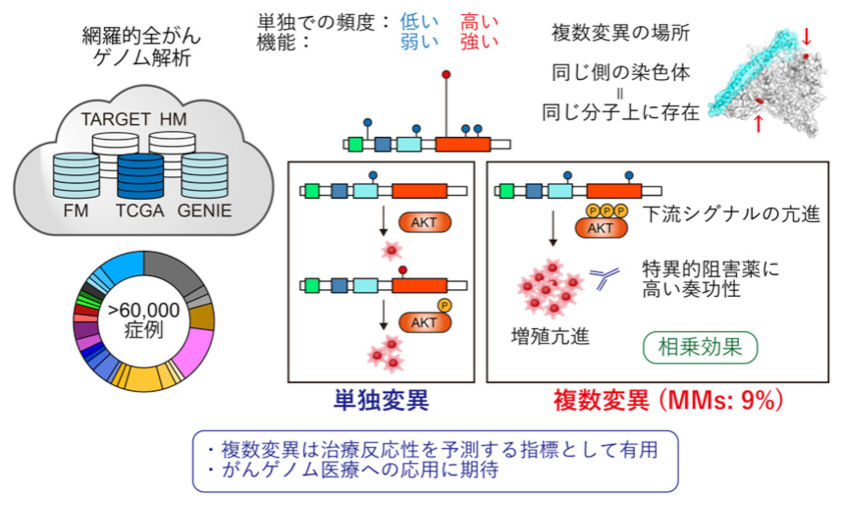
「横断的がんマルチオミクス解析による新規の発がん機構の解明」
我々は、これまで最大規模の症例数である6万例(150がん種以上)を超える大規模ながんゲノムデータについて、スーパーコンピューターを用いた遺伝子解析を行い、同一がん遺伝子内における複数変異が相乗的に機能するという新たな発がんメカニズムを解明しました (Y Saito, J Koya et al., Nature. 2020)。従来、がん遺伝子は単独で変異が生じることが多いと考えられてきましたが、一部のがん遺伝子では複数の変異が生じやすいことが明らかになりました。PIK3CA遺伝子・EGFR遺伝子などの代表的ながん遺伝子では変異を持つ症例の約10%が同一遺伝子内に複数の変異を有しており、これらの大部分は染色体の同じ側(シス)に起きていました。同一がん遺伝子に複数変異が生じる場合、単独の変異では低頻度でしか認められない部位やアミノ酸変化がより多く選択されていました。これらの変異は単独では機能的に弱い変異ですが、複数生じることで相乗効果により強い発がん促進作用を示しました。特にPIK3CA遺伝子で複数変異を持つ場合は、単独変異よりもより強い下流シグナルの活性化や当該遺伝子への依存度が認められ、特異的な阻害剤に対して感受性を示しました。これらの結果は、同一がん遺伝子内の複数変異が発がんに関与する新たな遺伝学的メカニズムであることを示しています。本研究により、これまで単独では意義不明であった変異が生じる理由が説明可能となるほか、複数変異は分子標的薬の治療反応性を予測するバイオマーカーにもなり得るため、がんゲノム診療に役立つことが期待されます。
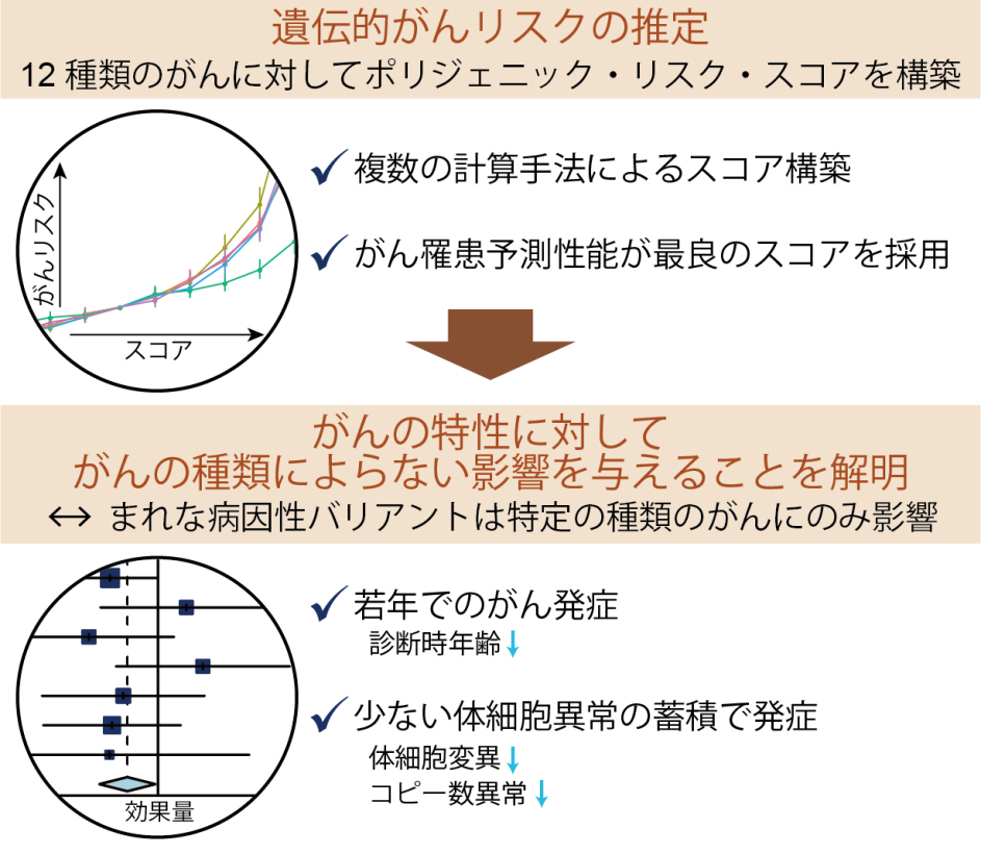
また、我々は体細胞異常に遺伝的素因が与える影響を理解するために、生殖細胞系列バリアントに着目した研究も進めています。がんの発症には、加齢・喫煙・放射線暴露など様々な「環境因子」が関与することが知られていますが、各個人の「遺伝因子」、すなわち「遺伝的がんリスク体質」も重要であることが知られています。近年、ゲノムワイド関連解析(GWAS解析)によって、多くの人が持つバリアントのうち数百~数千個ががんへのかかりやすさに影響を与えることが分かってきました。これらのバリアントを評価することにより、「がんになりやすい遺伝的な体質」(遺伝的がんリスク体質)を評価できると考えられます。
我々は、大阪大学の遺伝統計学教室(岡田随象教授)と共同で、様々な種類のがんに対して複数の計算手法を用いて各個人の「遺伝的がんリスク体質」を強く反映するスコアであるポリジェニック・リスク・スコア(PRS)を構築しました(Namba
S, Saito Y et al., Cancer Res.,
2022)。PRSを用いて遺伝的がんリスク体質の特性を網羅的に調べたところ、遺伝的がんリスク体質を持つ人は若い年齢でがんを発症し、がんに蓄積している体細胞異常(体細胞変異やコピー数異常)が少ないことが分かりました。また、これらの遺伝的がんリスク体質に関連する特性は、さまざまな種類のがんで共通して認められる事が分かりました。本研究成果によって「遺伝的がんリスク体質」の理解が進み、がんの予防や個別化医療を推進することに役立つと期待されます。
当研究室は、このように多様ながん種由来の様々な解析プラットフォームによる解析データを統合的に扱う、「横断的がんマルチオミクス解析」プロジェクトを進めており、重要な発がん機序の解明に取り組んでいます。
主要文献<研究テーマ(1)>
- Namba S, Saito Y (co-first), Kogure Y, et al. Common Germline Risk Variants Impact Somatic Alterations and Clinical Features across Cancers. Cancer Research, 83; 20-27, 2023. [36286845]
- Singaki S, Koya J, et al. Tumor-promoting function and regulatory landscape of PD-L2 in B-cell lymphoma. Leukemia, 2022. [36522456]
- Fukuhara S, Oshikawa-Kumade Y, Kogure Y, et al. Feasibility and clinical utility of comprehensive genomic profiling of hematological malignancies. Cancer Science, 113; 2763-2777,2022.[35579198]
- Kogure Y, Koya J (co-first), et al. Whole-genome landscape of adult T-cell leukemia/lymphoma. Blood, 139: 967-982, 2022. [34695199]
- Koya J, Saito Y (co-first), et al. Single-cell analysis of the multicellular ecosystem in viral carcinogenesis by HTLV-1. Blood Cancer Discov, 2:450-467, 2021. [34661162]
- Murakami K, Sasaki H, Nishiyama A, et al. A RUNX–CBFβ-driven enhancer directs the Irf8 dose-dependent lineage choice between DCs and monocytes. Nat Immunol, 22: 301-311, 2021. [33603226]
- Murakami K, Kurotaki D, Kawase W, et al. OGT Regulates Hematopoietic Stem Cell Maintenance via PINK1-Dependent Mitophagy. Cell Reports, 34: 108579, 2021 [33406421]
- Saito Y, Koya J (co-first), Araki M, et al. Landscape and function of multiple mutations within individual oncogenes. Nature, 582:95-99, 2020 [32494066]
- Kataoka K, Miyoshi H, Sakata S, et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia, 33:1687-1699, 2019 [30683910]
- Kataoka K, Iwanaga M, Yasunaga J, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood. 131:215-225, 2018 [29084771]
- Takeda Y, Kataoka K (co-first), Yamagishi J, et al. A TLR3-Specific Adjuvant Relieves Innate Resistance to PD-L1 Blockade without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep, 19:1874-1887, 2017 [28564605]
- Koya J, Kataoka K, Sato T, et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat Commun, 7:10924, 2016 [27010239]
- Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature, 534:402-406, 2016 [27281199]
- Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet, 47:1304-1315, 2015 [26437031]
- Kataoka K, Sato T, Yoshimi A, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med, 208:2403-2416, 2011 [22084405]
研究テーマ(2)
「造血幹細胞移植後の免疫再構築およびウイルス感染症に関する検討」
当院では、造血器疾患に対し、同種造血幹細胞移植を年間30-40例ほど行っています。造血幹細胞移植は唯一の根治療法ではありますが、大変リスクの高い治療でもあり、様々な移植後合併症を認め、治療に難渋することがあります。その一つとして、強力な化学療法と免疫抑制剤の影響で様々な感染症にかかりやすくなることが知られています。特にサイトメガロウイルス、ヒトヘルペスウイルス6、水痘帯状疱疹ウイルス、アデノウイルス、BKウイルスなどのウイルスは、時に命にかかわる重篤な感染症を引き起こします。 当研究室では、このようなウイルス感染症の予防および治療に結びつく知見を得ることを目的に、移植患者さんから同意を得て臨床検体を頂戴し、ウイルスに対する特異免疫や移植後免疫再構築について詳細の検討を行っています。 私たちは、患者さんのベッドサイドで生まれた疑問に基礎的にアプローチし、患者さんの実際の治療に還元することを目標に研究を進めています。
主要文献<研究テーマ(2)>
- Mori T, Kikuchi T, Kato J, et al. Seasonal changes in indoor airborne fungal concentration in a hematology ward. J Infect Chemother, 26:363-366, 2020 [31791593]
- Shiroshita K, Mori T, Kato J, et al. Clinical characteristics of human herpesvirus-6 myelitis after allogeneic hematopoietic stem cell transplantation and its favorable outcome by early intervention. Bone Marrow Transplant, 55:939-945, 2020 [31754252]
- Nakayama H, Yamazaki R, Mori T, et al. Human Herpesvirus 6 Reactivation Evaluated by Digital Polymerase Chain Reaction and Its Association With Dynamics of CD134-Positive T Cells After Allogeneic Hematopoietic Stem Cell Transplantation. J Infect Dis, 220:1001-1007, 2019 [31063196]
- Yamazaki R, Kikuchi T, Mori T, et al. Recurrent bacterial pneumonia due to immunoglobulin G2 subclass deficiency after allogeneic hematopoietic stem cell transplantation: Efficacy of immunoglobulin replacement. Transpl Infect Dis, 20:e12863, 2018 [30929295]
- Kato J, Mori T, Suzuki T, et al. Nosocomial BK Polyomavirus infection causing hemorrhagic cystitis among patients with hematological malignancies after hematopoietic stem cell transplantation. Am J Transplant, 17:2428-2433, 2017 [28295968]
今後の展望
現在は、造血器腫瘍を中心に、未だに遺伝子異常が十分に明らかではない悪性腫瘍の網羅的な遺伝子解析を行うだけでなく、我々がPD-L1 3′-UTR異常を同定したのと同様のがん横断的なアプローチにより、悪性腫瘍全体の遺伝子異常の全体像を明らかにしようと試みています。特に、次世代シーケンスにより得られたビッグデータを医学的・生物学的に異なる視点から解析・解釈することにより、新たな発見を見出すことを目標としています。同時に、新規に同定された遺伝子異常が腫瘍化に果たす役割について、CRISPR/Cas9システムなどの最先端の分子生物学的手法や遺伝子改変動物モデルなどを用いて解明することを目指しています。さらに、このような研究成果を臨床に還元するために、遺伝子異常に対する分子標的薬の開発やバイオマーカーとしての意義の検討や、造血器腫瘍に対する遺伝子解析パネルの開発、臨床シーケンスなどを含めた個別化医療への応用に取り組んでいます。
注意事項:記述、画像、写真の無断転載・転用を禁止します
大学院生がお世話になっている研究室
国立国際医療研究センター研究所生体恒常性プロジェクト(田久保圭誉 生体恒常性プロジェクト長)
綿貫慎太郎、城下郊平、藤田進也
東京大学医科学研究所幹細胞治療研究センター幹細胞分子医学分野(岩間厚志 教授)
神谷高博
